Problem 01
Strongly correlated metalloclusters and catalytic materials
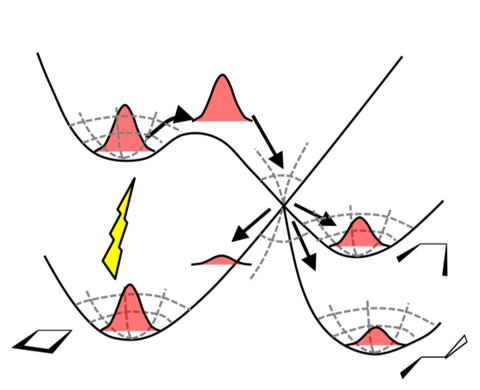
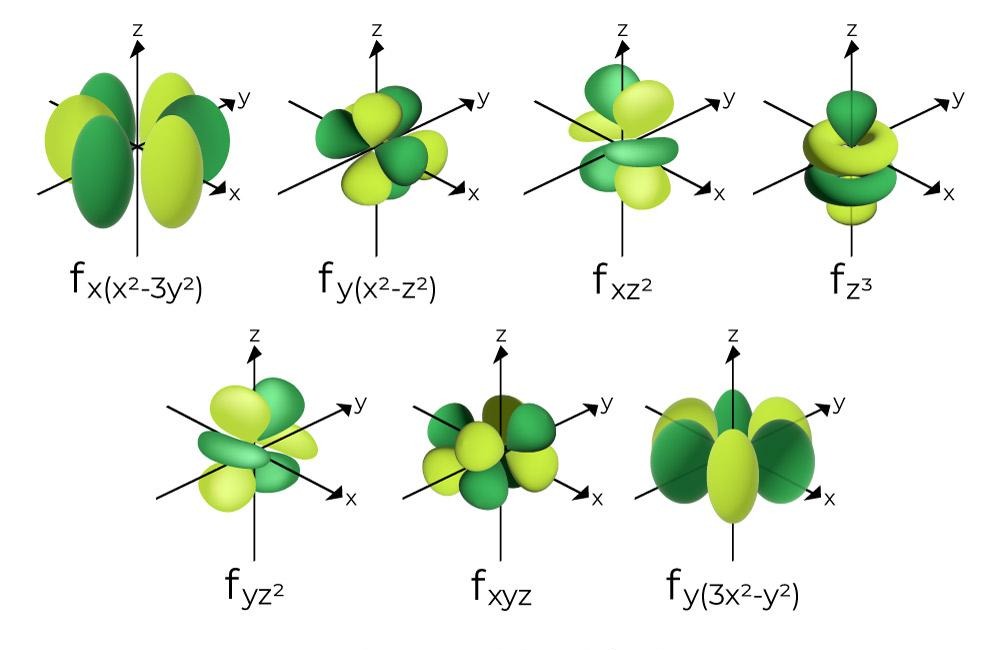
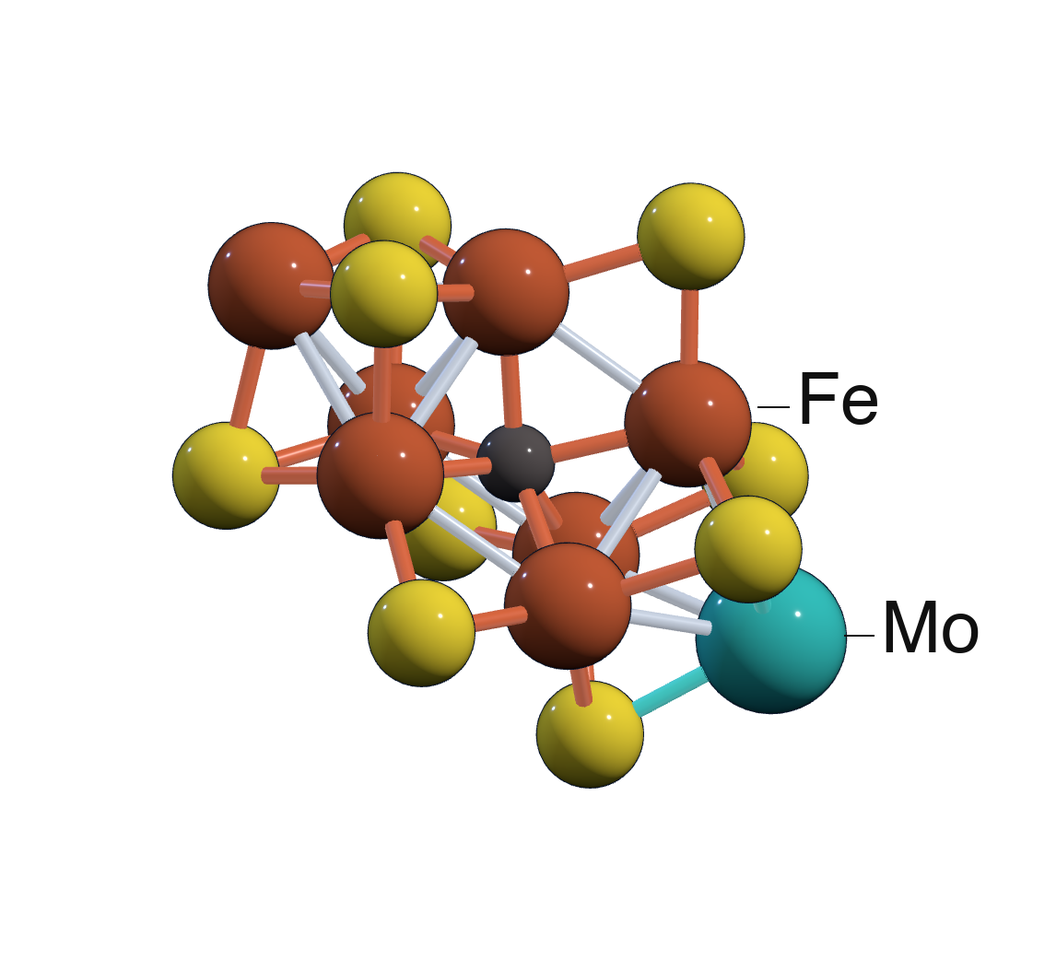
Strongly correlated systems such as FeMoco challenge quantum-chemistry methods. These systems involve high-rank excitations and require accurate control of large Hilbert spaces to reach chemically meaningful energy scales.
Image: Asthana Group.